Day - 1 Introduction to R and RStudio
1.1 Learning Objectives
By the end of Day 1, you will be able to:
- Install and navigate RStudio effectively
- Understand basic R data structures (vectors, data frames, lists)
- Import and explore simple datasets
- Write basic control flow structures and functions
1.2 Module 1: Setting Up and Getting Started with R
1.2.1 Introduction
R is a powerful programming language and environment specifically designed for statistical computing and graphics. RStudio is an integrated development environment (IDE) that makes working with R much easier.
1.2.2 Installing R and RStudio
-
Install R (version 4.3.0 or higher)
- Visit https://cran.r-project.org/
- Download the version appropriate for your operating system
- Run the installer
-
Install RStudio Desktop
- Visit https://posit.co/download/rstudio-desktop/
- Download the free Desktop version
- Run the installer
-
Install Required R Packages
Open an R session in the terminal or RStudio:
# Install CRAN packages install.packages(c( "bookdown", "rmarkdown", "knitr", "pheatmap", "ggplot2", "downlit", "xml2", "reshape2", "gridExtra", "tidyverse", "lme4", "ggforce", "scatterpie", "png" # Optional )) # Install Bioconductor packages if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install(c( "limma", "vsn", "sva", "clusterProfiler", "org.Hs.eg.db", "lme4", "KEGGREST", "AnnotationDbi", "annotate", "GO.db", "genefilter", "GOSemSim", "DOSE", "enrichplot" ), update = TRUE, ask = FALSE)
1.2.3 RStudio Interface Tour
RStudio has four main panes:
- Source Editor (top-left): Where you write and edit your scripts
- Console (bottom-left): Where code is executed and results appear
- Environment/History (top-right): Shows objects in memory and command history
- Files/Plots/Packages/Help (bottom-right): File browser, plot viewer, package manager, and help documentation
1.2.4 Scripts vs Console
The Console is for: - Quick calculations - Testing commands - Interactive exploration
Scripts (.R or .Rmd files) are for: - Saving your work - Creating reproducible analyses - Organizing complex workflows
1.2.5 Basic Operators
# Arithmetic operators
5 + 3 # Addition
#> [1] 8
10 - 4 # Subtraction
#> [1] 6
6 * 7 # Multiplication
#> [1] 42
20 / 4 # Division
#> [1] 5
2 ^ 3 # Exponentiation
#> [1] 8
17 %% 5 # Modulo (remainder)
#> [1] 2
# Comparison operators
5 == 5 # Equal to
#> [1] TRUE
5 != 3 # Not equal to
#> [1] TRUE
7 > 3 # Greater than
#> [1] TRUE
4 < 8 # Less than
#> [1] TRUE
5 >= 5 # Greater than or equal
#> [1] TRUE
3 <= 10 # Less than or equal
#> [1] TRUE
# Logical operators
TRUE & FALSE # AND
#> [1] FALSE
TRUE | FALSE # OR
#> [1] TRUE
!TRUE # NOT
#> [1] FALSE
# Scientific notation
1.5e6 # 1,500,000
#> [1] 1500000Tip: Use descriptive variable names with underscores
1.2.6 Variables & Assignment
# Assignment operator
x <- 10 # Assign 10 to x
y = 5 # Alternative (but <- is preferred)
## Assign values to variables
protein_count <- 800
sample_size <- 12
study_name <- "Proteomics_2025"
## View variables
protein_count
#> [1] 800
sample_size
#> [1] 12
## Use variables in calculations
total_measurements <- protein_count * sample_size
total_measurements
#> [1] 96001.2.7 Data Types
# Numeric
intensity <- 25114306.44
class(intensity)
#> [1] "numeric"
# Character (text)
accession <- "F1LMU0"
class(accession)
#> [1] "character"
# Logical
is_significant <- TRUE
class(is_significant)
#> [1] "logical"
# Check type
is.numeric(intensity)
#> [1] TRUE
is.character(accession)
#> [1] TRUE1.2.8 Creating Your First Script
# Create a new R script: File > New File > R Script
# Or use Ctrl+Shift+N (Windows/Linux) or Cmd+Shift+N (Mac)
# Write your code
message("Hello, Proteomics World!")
# Save your script: File > Save
# Run code: Ctrl+Enter (Windows/Linux) or Cmd+Return (Mac)1.3 Module 2: Data Types and Structures
1.3.1 Vectors
Vectors are the most basic data structure in R. They contain elements of the same type.
# Numeric vectors
ages <- c(25, 30, 35, 40, 45)
print(ages)
#> [1] 25 30 35 40 45
# Character vectors
names <- c("Alice", "Bob", "Charlie", "Diana", "Eve")
print(names)
#> [1] "Alice" "Bob" "Charlie" "Diana" "Eve"
# Logical vectors
passed_qc <- c(TRUE, TRUE, FALSE, TRUE, TRUE)
print(passed_qc)
#> [1] TRUE TRUE FALSE TRUE TRUE
# Sequences
seq_1_10 <- 1:10
seq_custom <- seq(from = 0, to = 100, by = 10)
# Vector operations
mean(ages)
#> [1] 351.3.2 Indexing and Subsetting
# Access elements by position (1-indexed!)
ages[1] # First element
#> [1] 25
ages[c(1, 3, 5)] # Multiple elements
#> [1] 25 35 45
ages[-2] # All except second element
#> [1] 25 35 40 45
# Logical indexing
ages[ages > 35] # Elements greater than 35
#> [1] 40 45
# Named vectors
protein_abundance <- c(ACTB = 1500, GAPDH = 2000, MYC = 800)
protein_abundance["ACTB"]
#> ACTB
#> 15001.3.3 Data Frames
Data frames are the most common structure for storing tabular data.
# Create a data frame
patient_data <- data.frame(
patient_id = 1:5,
name = c("Alice", "Bob", "Charlie", "Diana", "Eve"),
age = c(25, 30, 35, 40, 45),
treatment = c("A", "B", "A", "B", "A"),
response = c(TRUE, TRUE, FALSE, TRUE, FALSE),
stringsAsFactors = FALSE
)
print(patient_data)
#> patient_id name age treatment response
#> 1 1 Alice 25 A TRUE
#> 2 2 Bob 30 B TRUE
#> 3 3 Charlie 35 A FALSE
#> 4 4 Diana 40 B TRUE
#> 5 5 Eve 45 A FALSE
# View structure
str(patient_data)
#> 'data.frame': 5 obs. of 5 variables:
#> $ patient_id: int 1 2 3 4 5
#> $ name : chr "Alice" "Bob" "Charlie" "Diana" ...
#> $ age : num 25 30 35 40 45
#> $ treatment : chr "A" "B" "A" "B" ...
#> $ response : logi TRUE TRUE FALSE TRUE FALSE
# Summary statistics
summary(patient_data)
#> patient_id name age
#> Min. :1 Length:5 Min. :25
#> 1st Qu.:2 Class :character 1st Qu.:30
#> Median :3 Mode :character Median :35
#> Mean :3 Mean :35
#> 3rd Qu.:4 3rd Qu.:40
#> Max. :5 Max. :45
#> treatment response
#> Length:5 Mode :logical
#> Class :character FALSE:2
#> Mode :character TRUE :3
#>
#>
#>
# Access columns
patient_data$age
#> [1] 25 30 35 40 45
patient_data[, "name"]
#> [1] "Alice" "Bob" "Charlie" "Diana" "Eve"
patient_data[, 2]
#> [1] "Alice" "Bob" "Charlie" "Diana" "Eve"
# Access rows
patient_data[1, ] # First row
#> patient_id name age treatment response
#> 1 1 Alice 25 A TRUE
patient_data[1:3, ] # First three rows
#> patient_id name age treatment response
#> 1 1 Alice 25 A TRUE
#> 2 2 Bob 30 B TRUE
#> 3 3 Charlie 35 A FALSE
# Access specific cells
patient_data[2, 3] # Row 2, Column 3
#> [1] 30
patient_data[2, "age"] # Same, using column name
#> [1] 30
# Subset by condition
patient_data[patient_data$age > 30, ]
#> patient_id name age treatment response
#> 3 3 Charlie 35 A FALSE
#> 4 4 Diana 40 B TRUE
#> 5 5 Eve 45 A FALSE
patient_data[patient_data$treatment == "A", ]
#> patient_id name age treatment response
#> 1 1 Alice 25 A TRUE
#> 3 3 Charlie 35 A FALSE
#> 5 5 Eve 45 A FALSE1.3.3.1 Exploring Data Frames
# View first rows
head(patient_data, 2)
#> patient_id name age treatment response
#> 1 1 Alice 25 A TRUE
#> 2 2 Bob 30 B TRUE
# Dimensions
dim(patient_data)
#> [1] 5 5
nrow(patient_data)
#> [1] 5
ncol(patient_data)
#> [1] 5
# Column names
colnames(patient_data)
#> [1] "patient_id" "name" "age" "treatment"
#> [5] "response"1.3.3.2 Accessing Data Frame Elements
# Access column by name
patient_data$name
#> [1] "Alice" "Bob" "Charlie" "Diana" "Eve"
# Access using brackets [row, column]
patient_data[1, 3] # Row 1, column 3
#> [1] 25
patient_data[1:2, ] # First 2 rows, all columns
#> patient_id name age treatment response
#> 1 1 Alice 25 A TRUE
#> 2 2 Bob 30 B TRUE
patient_data[, "treatment"] # All rows, Mass column
#> [1] "A" "B" "A" "B" "A"
# Subset data
patient_data[patient_data$age > 30, ]
#> patient_id name age treatment response
#> 3 3 Charlie 35 A FALSE
#> 4 4 Diana 40 B TRUE
#> 5 5 Eve 45 A FALSE1.3.5 Factors
Factors are used for categorical data.
# Create factor
treatment_factor <- factor(c("Control", "Drug A", "Drug B", "Control", "Drug A"))
print(treatment_factor)
#> [1] Control Drug A Drug B Control Drug A
#> Levels: Control Drug A Drug B
# Check levels
levels(treatment_factor)
#> [1] "Control" "Drug A" "Drug B"
# Ordered factors
severity <- factor(
c("Mild", "Severe", "Moderate", "Mild", "Severe"),
levels = c("Mild", "Moderate", "Severe"),
ordered = TRUE
)
print(severity)
#> [1] Mild Severe Moderate Mild Severe
#> Levels: Mild < Moderate < Severe1.3.6 Type Coercion
# Implicit coercion
mixed <- c(1, 2, "three", 4) # All converted to character
print(mixed)
#> [1] "1" "2" "three" "4"
# Explicit coercion
numbers_char <- c("1", "2", "3", "4")
numbers_num <- as.numeric(numbers_char)
print(numbers_num)
#> [1] 1 2 3 4
# Check types
class(mixed)
#> [1] "character"
is.numeric(mixed)
#> [1] FALSE
is.character(mixed)
#> [1] TRUE1.3.7 Exercise 1.2: Data Structures
Create a data frame for a proteomic experiment with:
- 10 protein IDs (P001 to P010)
- Random abundance values between 100 and 5000
- Random p-values between 0 and 1
- Significance status (TRUE if p-value < 0.05)
# Solution
set.seed(42) # For reproducibility
proteins <- data.frame(
protein_id = paste0("P", sprintf("%03d", 1:10)),
abundance = round(runif(10, min = 100, max = 5000), 2),
p_value = runif(10, min = 0, max = 1),
stringsAsFactors = FALSE
)
proteins$significant <- proteins$p_value < 0.05
print(proteins)
#> protein_id abundance p_value significant
#> 1 P001 4582.55 0.4577418 FALSE
#> 2 P002 4691.67 0.7191123 FALSE
#> 3 P003 1502.08 0.9346722 FALSE
#> 4 P004 4169.19 0.2554288 FALSE
#> 5 P005 3244.55 0.4622928 FALSE
#> 6 P006 2643.57 0.9400145 FALSE
#> 7 P007 3709.28 0.9782264 FALSE
#> 8 P008 759.87 0.1174874 FALSE
#> 9 P009 3319.26 0.4749971 FALSE
#> 10 P010 3554.82 0.5603327 FALSE
# Summary
cat("\nNumber of significant proteins:", sum(proteins$significant), "\n")
#>
#> Number of significant proteins: 01.4 Module 3: Control Flow and Functions
1.4.1 Conditional Statements
# if statement
x <- 10
if (x > 5) {
print("x is greater than 5")
}
#> [1] "x is greater than 5"
# if-else
if (x > 15) {
print("x is greater than 15")
} else {
print("x is 15 or less")
}
#> [1] "x is 15 or less"
# if-else if-else
score <- 75
if (score >= 90) {
grade <- "A"
} else if (score >= 80) {
grade <- "B"
} else if (score >= 70) {
grade <- "C"
} else {
grade <- "F"
}
print(paste("Grade:", grade))
#> [1] "Grade: C"
# Vectorized ifelse
values <- c(1, 5, 10, 15, 20)
categories <- ifelse(values > 10, "High", "Low")
print(categories)
#> [1] "Low" "Low" "Low" "High" "High"1.4.2 Loops
# for loop
for (i in 1:5) {
print(paste("Iteration:", i))
}
#> [1] "Iteration: 1"
#> [1] "Iteration: 2"
#> [1] "Iteration: 3"
#> [1] "Iteration: 4"
#> [1] "Iteration: 5"
# Loop through vector
proteins <- c("ACTB", "GAPDH", "MYC")
for (protein in proteins) {
print(paste("Processing:", protein))
}
#> [1] "Processing: ACTB"
#> [1] "Processing: GAPDH"
#> [1] "Processing: MYC"
# while loop
counter <- 1
while (counter <= 5) {
print(paste("Counter:", counter))
counter <- counter + 1
}
#> [1] "Counter: 1"
#> [1] "Counter: 2"
#> [1] "Counter: 3"
#> [1] "Counter: 4"
#> [1] "Counter: 5"
# Loop with condition
numbers <- 1:10
for (num in numbers) {
if (num %% 2 == 0) {
print(paste(num, "is even"))
}
}
#> [1] "2 is even"
#> [1] "4 is even"
#> [1] "6 is even"
#> [1] "8 is even"
#> [1] "10 is even"1.4.3 Functions
# Basic function
greet <- function(name) {
message <- paste("Hello,", name, "!")
return(message)
}
greet("Alice")
#> [1] "Hello, Alice !"
# Function with multiple parameters
calculate_fold_change <- function(treatment, control) {
fc <- treatment / control
log2_fc <- log2(fc)
return(log2_fc)
}
calculate_fold_change(treatment = 200, control = 100)
#> [1] 1
# Function with default parameters
normalize_abundance <- function(abundance, method = "median") {
if (method == "median") {
normalized <- abundance / median(abundance, na.rm = TRUE)
} else if (method == "mean") {
normalized <- abundance / mean(abundance, na.rm = TRUE)
} else {
stop("Method must be 'median' or 'mean'")
}
return(normalized)
}
values <- c(100, 200, 300, 400, 500)
normalize_abundance(values)
#> [1] 0.3333333 0.6666667 1.0000000 1.3333333 1.6666667
normalize_abundance(values, method = "mean")
#> [1] 0.3333333 0.6666667 1.0000000 1.3333333 1.66666671.4.4 Apply Family Functions
# Create sample data
protein_matrix <- matrix(
c(100, 150, 200, 250,
110, 160, 210, 260,
120, 170, 220, 270),
nrow = 3, byrow = TRUE
)
colnames(protein_matrix) <- c("Sample1", "Sample2", "Sample3", "Sample4")
rownames(protein_matrix) <- c("Protein1", "Protein2", "Protein3")
print(protein_matrix)
#> Sample1 Sample2 Sample3 Sample4
#> Protein1 100 150 200 250
#> Protein2 110 160 210 260
#> Protein3 120 170 220 270
# apply: apply function to rows or columns
row_means <- apply(protein_matrix, 1, mean) # 1 = rows
col_means <- apply(protein_matrix, 2, mean) # 2 = columns
print(row_means)
#> Protein1 Protein2 Protein3
#> 175 185 195
print(col_means)
#> Sample1 Sample2 Sample3 Sample4
#> 110 160 210 260
# lapply: apply function to list, returns list
my_list <- list(a = 1:5, b = 6:10, c = 11:15)
list_means <- lapply(my_list, mean)
print(list_means)
#> $a
#> [1] 3
#>
#> $b
#> [1] 8
#>
#> $c
#> [1] 13
# sapply: simplified version of lapply
vector_means <- sapply(my_list, mean)
print(vector_means)
#> a b c
#> 3 8 131.4.5 Exercise 1.3: Functions and Loops
Write a function that:
- Takes a vector of protein abundances
- Calculates the coefficient of variation (CV = sd/mean * 100)
- Returns “Pass” if CV < 20%, “Fail” otherwise
Apply this function to multiple samples using a loop.
# Solution
calculate_cv_status <- function(abundances) {
cv <- (sd(abundances, na.rm = TRUE) / mean(abundances, na.rm = TRUE)) * 100
if (cv < 20) {
status <- "Pass"
} else {
status <- "Fail"
}
return(list(cv = round(cv, 2), status = status))
}
# Create sample data
sample_data <- list(
sample1 = c(100, 105, 98, 102, 99),
sample2 = c(100, 150, 90, 200, 80),
sample3 = c(500, 505, 498, 502, 496)
)
# Apply function
for (sample_name in names(sample_data)) {
result <- calculate_cv_status(sample_data[[sample_name]])
cat(sample_name, "- CV:", result$cv, "% - Status:", result$status, "\n")
}
#> sample1 - CV: 2.75 % - Status: Pass
#> sample2 - CV: 40.56 % - Status: Fail
#> sample3 - CV: 0.7 % - Status: Pass
1.5 Module 4: Data Visualization with ggplot2
In this module, we will explore how to visualize data using ggplot2, one of the most powerful and flexible plotting systems in R.
1.5.2 Example Dataset
We’ll first create an example dataset representing protein abundance under two conditions: Control and Treatment.
set.seed(42)
# Simulate realistic protein abundance data
n_proteins <- 100
protein_data <- data.frame(
protein_id = paste0("P", sprintf("%03d", 1:n_proteins)),
abundance = c(
rnorm(n_proteins / 2, mean = 1000, sd = 150), # Control group
rnorm(n_proteins / 2, mean = 1300, sd = 180) # Treatment group
),
condition = rep(c("Control", "Treatment"), each = n_proteins / 2)
)
# Add a few outliers to make the data more realistic
outlier_indices <- sample(1:n_proteins, 3)
protein_data$abundance[outlier_indices] <-
protein_data$abundance[outlier_indices] * runif(3, 1.5, 2)
head(protein_data)
#> protein_id abundance condition
#> 1 P001 1205.6438 Control
#> 2 P002 915.2953 Control
#> 3 P003 1054.4693 Control
#> 4 P004 1094.9294 Control
#> 5 P005 1060.6402 Control
#> 6 P006 984.0813 Control1.5.3 Histogram: Distribution of Protein Abundance
A histogram helps visualize the distribution of continuous variables such as protein abundance.
ggplot(protein_data, aes(x = abundance)) +
geom_histogram(binwidth = 100, fill = "steelblue", color = "white") +
labs(
title = "Distribution of Protein Abundance",
x = "Abundance",
y = "Count"
) +
theme_minimal()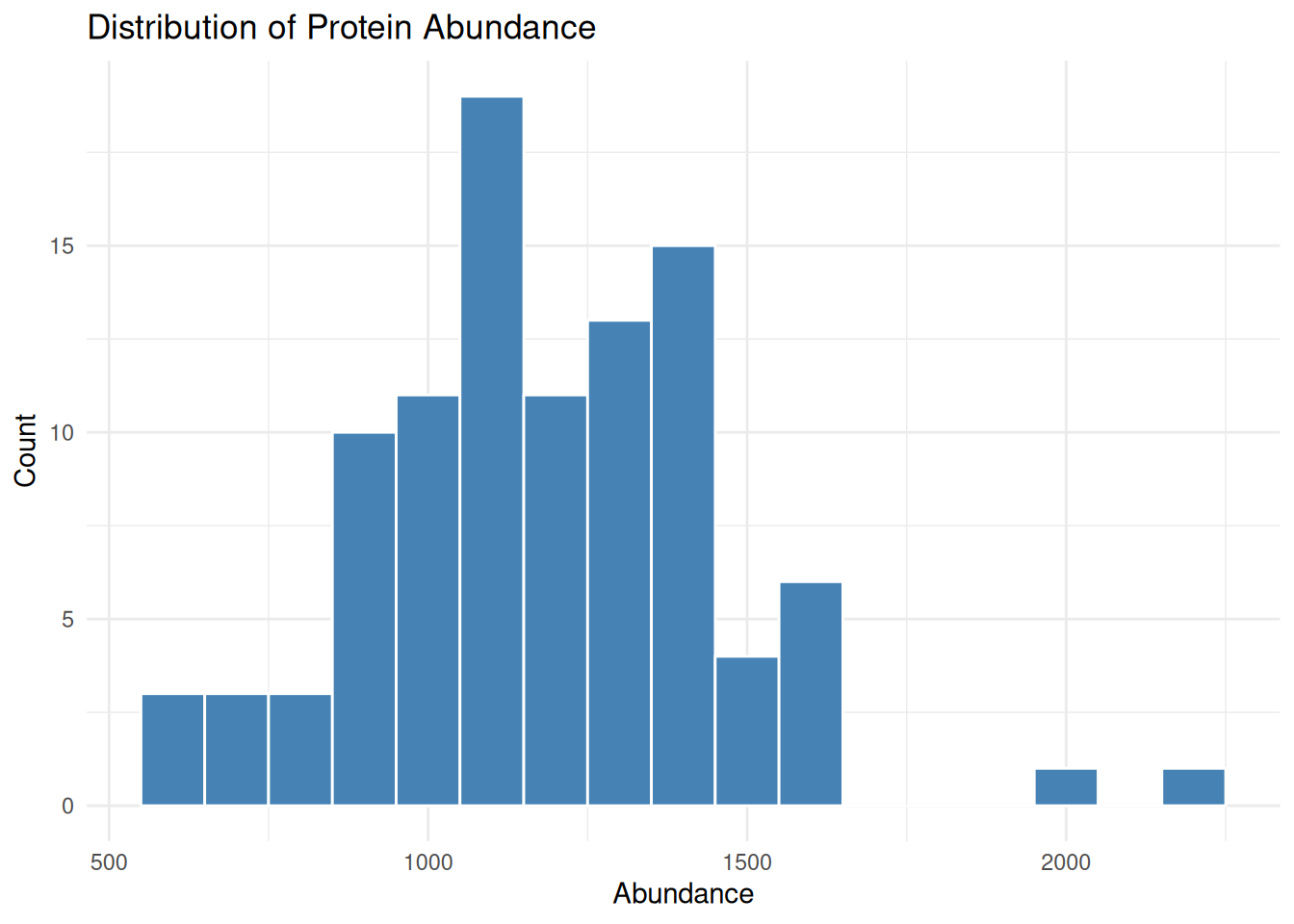
1.5.4 Boxplot: Comparing Conditions
A boxplot provides a compact summary of abundance values across conditions.
ggplot(protein_data, aes(x = condition, y = abundance, fill = condition)) +
geom_boxplot(alpha = 0.7) +
labs(
title = "Protein Abundance by Condition",
x = "Condition",
y = "Abundance"
) +
theme_minimal() +
theme(legend.position = "none")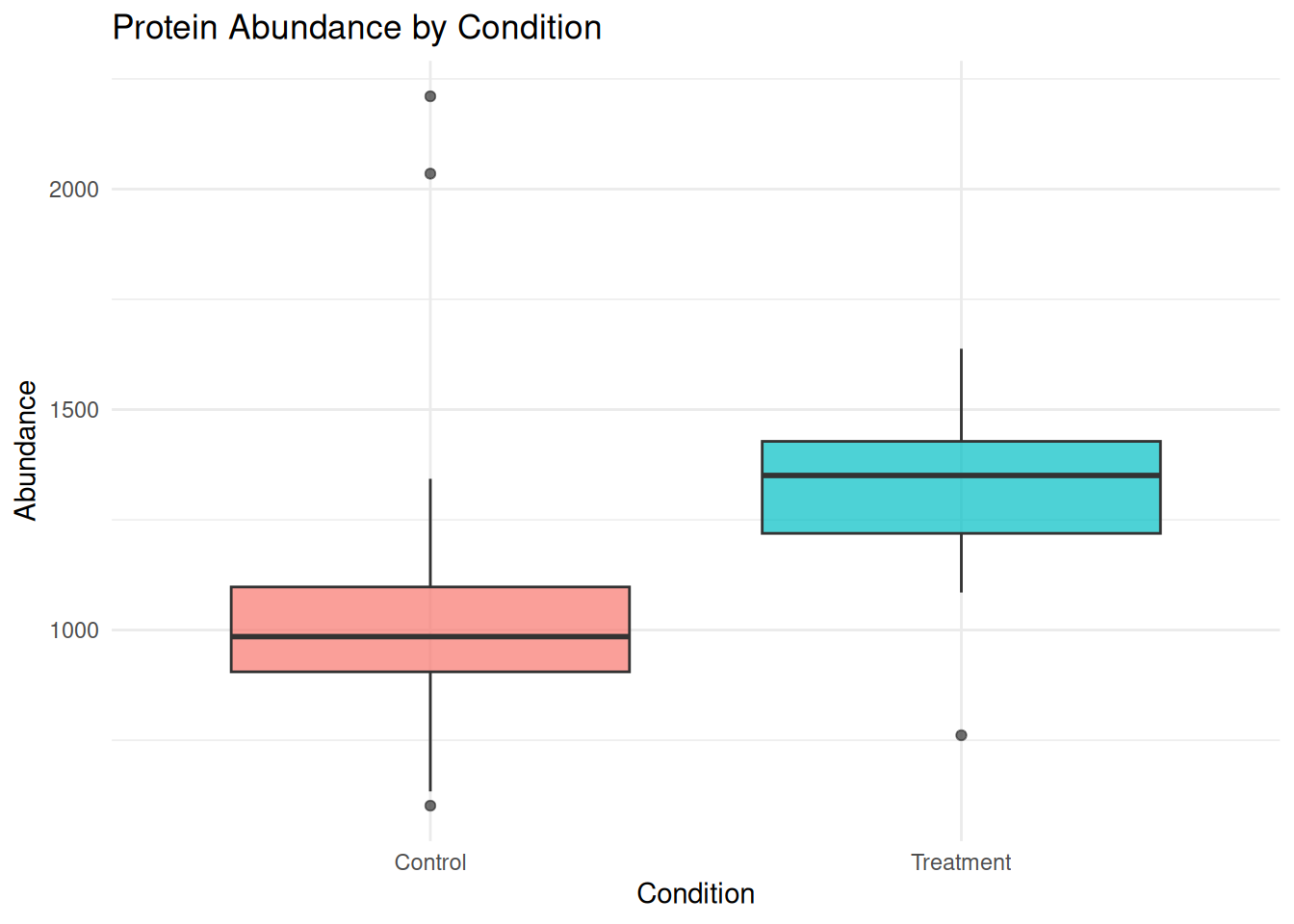
1.5.5 Scatter Plot: Individual Points with Mean Values
A scatter plot with jittering can display individual data points while avoiding overlap. Adding mean points provides an overview of group averages.
ggplot(protein_data, aes(x = condition, y = abundance, color = condition)) +
geom_jitter(width = 0.2, alpha = 0.6) +
stat_summary(fun = mean, geom = "point", size = 4, shape = 18, color = "black") +
labs(
title = "Abundance per Condition with Mean Values",
x = "Condition",
y = "Abundance"
) +
theme_minimal()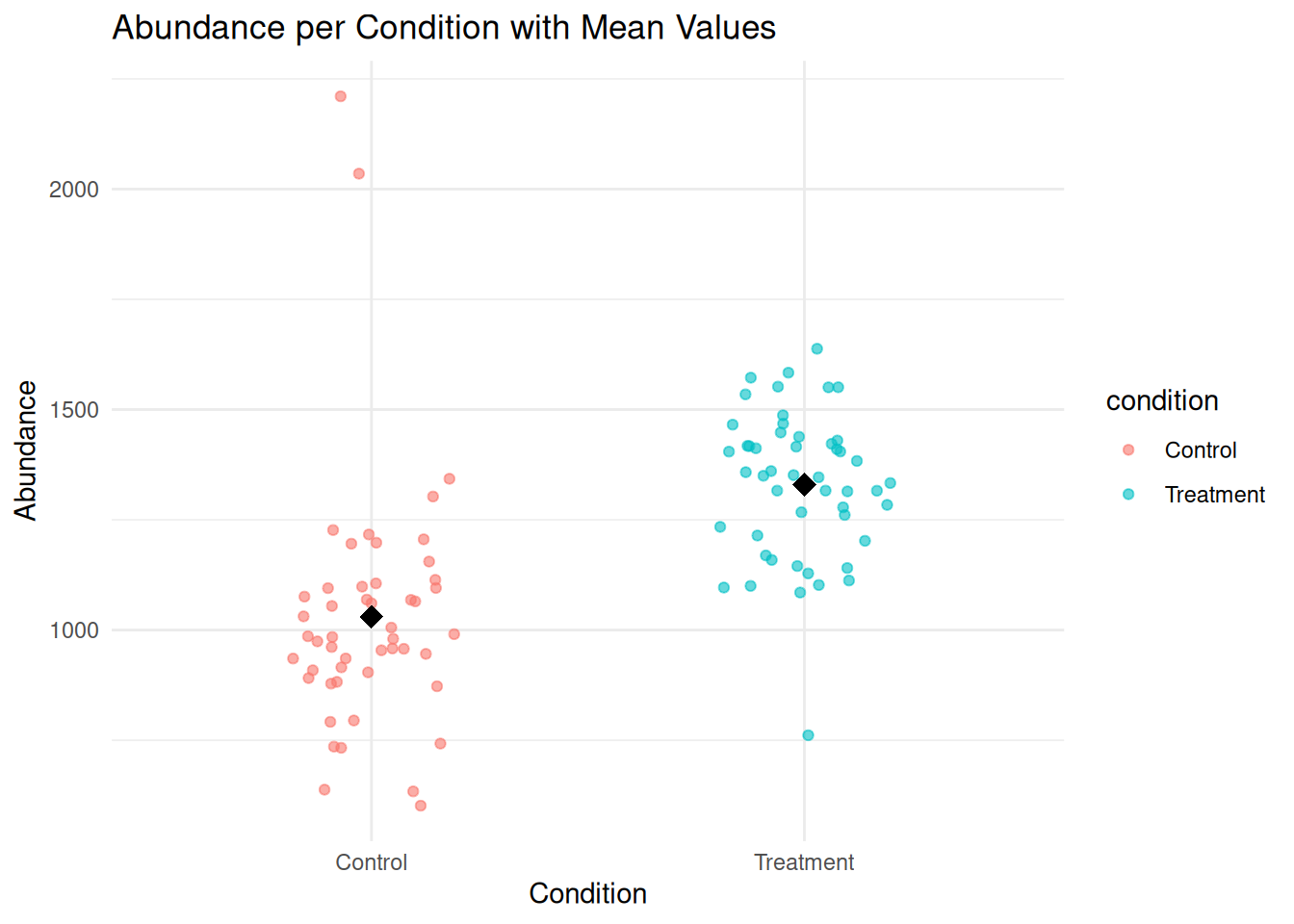
1.5.6 Density Plot: Distribution by Condition
A density plot shows the smoothed distribution of abundance values for each condition.
ggplot(protein_data, aes(x = abundance, fill = condition)) +
geom_density(alpha = 0.5) +
labs(
title = "Density Plot of Protein Abundance by Condition",
x = "Abundance",
y = "Density"
) +
theme_minimal()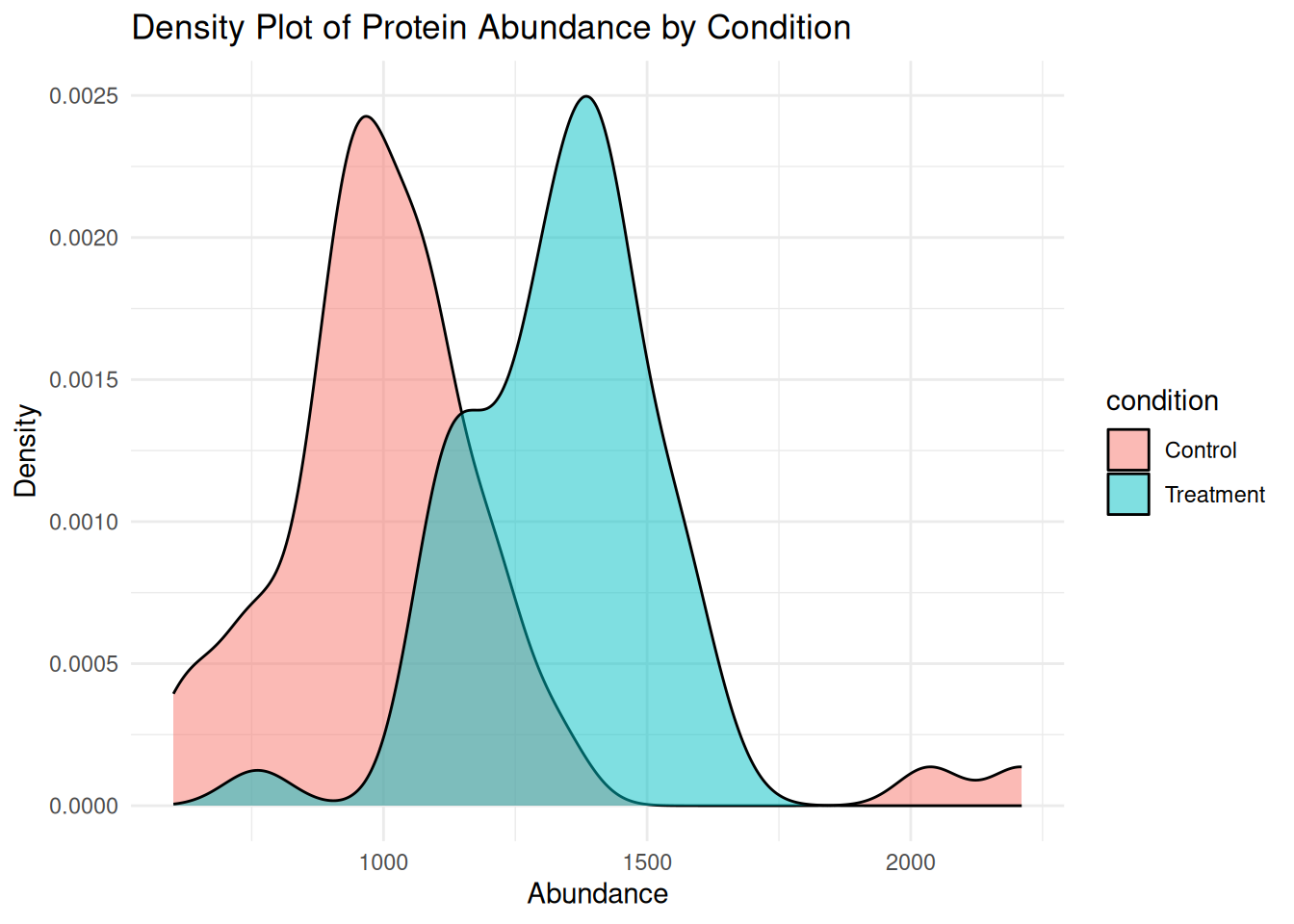
1.5.7 Summary
In this module, you learned how to:
- Visualize distributions using histograms and density plots
- Compare groups using boxplots
- Display individual data points with jitter and summary statistics
These visualizations are essential for exploring proteomics data and identifying trends or outliers before further statistical analysis.
1.6 Module 5: Importing and Exploring Data
1.6.1 Reading CSV Files
# Read CSV
data <- read.csv("data/proteins.csv")
# Read with tidyverse
library(readr)
data <- read_csv("data/proteins.csv")
# Read tab-delimited
data <- read.delim("data/proteins.txt", sep = "\t")1.6.2 Basic Data Exploration
# Create example data
set.seed(123)
protein_data <- data.frame(
protein_id = paste0("P", 1:100),
abundance = rnorm(100, mean = 1000, sd = 200),
condition = rep(c("Control", "Treatment"), each = 50)
)
# Dimensions
dim(protein_data)
#> [1] 100 3
nrow(protein_data)
#> [1] 100
ncol(protein_data)
#> [1] 3
# First and last rows
head(protein_data)
#> protein_id abundance condition
#> 1 P1 887.9049 Control
#> 2 P2 953.9645 Control
#> 3 P3 1311.7417 Control
#> 4 P4 1014.1017 Control
#> 5 P5 1025.8575 Control
#> 6 P6 1343.0130 Control
tail(protein_data)
#> protein_id abundance condition
#> 95 P95 1272.1305 Treatment
#> 96 P96 879.9481 Treatment
#> 97 P97 1437.4666 Treatment
#> 98 P98 1306.5221 Treatment
#> 99 P99 952.8599 Treatment
#> 100 P100 794.7158 Treatment
# Summary statistics
summary(protein_data)
#> protein_id abundance condition
#> Length:100 Min. : 538.2 Length:100
#> Class :character 1st Qu.: 901.2 Class :character
#> Mode :character Median :1012.4 Mode :character
#> Mean :1018.1
#> 3rd Qu.:1138.4
#> Max. :1437.5
# Table for categorical data
table(protein_data$condition)
#>
#> Control Treatment
#> 50 501.7 Module 6: Data Wrangling with tidyverse
1.7.1 Introduction to tidyverse
What is tidyverse?
- Collection of R packages for data science
- Consistent syntax and design philosophy
- Main packages: dplyr, tidyr, ggplot2, readr
Why tidyverse?
- Readable, intuitive code
- Pipe operator
%>%for chaining operations - Faster learning curve
- Industry standard
1.7.2 The Pipe Operator %>%
Traditional approach:
With pipe operator:
Read as: “Take x, THEN take square root, THEN take log10, THEN calculate mean”
1.7.3 dplyr: Grammar of Data Manipulation
Key Functions:
-
select()- Choose columns -
filter()- Choose rows based on conditions -
mutate()- Create or modify columns -
arrange()- Sort data -
summarize()- Calculate summary statistics -
group_by()- Group data for operations
1.7.4 Your Actual Proteomics Data Structure
# Simulate your data structure
set.seed(123)
proteomics_data <- readxl::read_excel(
path = "examples/2126001_Protein Export_ex.xlsx", #this should the file example that genethon provided
skip = 1
)
proteomics_data <- proteomics_data %>%
select(where(~ any(!is.na(.))))
head(proteomics_data[, 1:10])
#> # A tibble: 6 × 10
#> Accession Gene `Peptide count` `Unique peptides`
#> <chr> <chr> <dbl> <dbl>
#> 1 F1LMU0 F1LMU0 338 138
#> 2 G3V8V3 G3V8V3 146 113
#> 3 A0A0G2JSP8 A0A0G2JSP8 117 109
#> 4 Q64578 AT2A1 126 89
#> 5 D4AEH9 D4AEH9 87 83
#> 6 P15429 ENOB 110 83
#> # ℹ 6 more variables: `Confidence score` <dbl>, Mass <dbl>,
#> # Description <chr>, `2126001_029_F5` <dbl>,
#> # `2126001_359_F9` <dbl>, `2126001_401_F1` <dbl>Good practice: Convert all column names to lowercase and replace spaces with underscores _, to keep the naming of colmuns cleaner and more consistent.
#colnames(proteomics_data) <- tolower(colnames(proteomics_data))
colnames(proteomics_data) <- gsub(" ", "_", tolower(colnames(proteomics_data)))1.7.5 select(): Choose Columns
# Select specific columns
proteomics_data %>%
select(accession, gene, mass, `2126001_029_f5`) %>%
head(3)
#> # A tibble: 3 × 4
#> accession gene mass `2126001_029_f5`
#> <chr> <chr> <dbl> <dbl>
#> 1 F1LMU0 F1LMU0 222850. 25114306.
#> 2 G3V8V3 G3V8V3 97288. 18519365.
#> 3 A0A0G2JSP8 A0A0G2JSP8 43019. 897562438.
# Select range of columns
proteomics_data %>%
select(accession:mass) %>%
head(3)
#> # A tibble: 3 × 6
#> accession gene peptide_count unique_peptides
#> <chr> <chr> <dbl> <dbl>
#> 1 F1LMU0 F1LMU0 338 138
#> 2 G3V8V3 G3V8V3 146 113
#> 3 A0A0G2JSP8 A0A0G2JSP8 117 109
#> # ℹ 2 more variables: confidence_score <dbl>, mass <dbl>
# Select columns containing "sample"
proteomics_data %>%
select(accession, contains("f5")) %>%
head(3)
#> # A tibble: 3 × 2
#> accession `2126001_029_f5`
#> <chr> <dbl>
#> 1 F1LMU0 25114306.
#> 2 G3V8V3 18519365.
#> 3 A0A0G2JSP8 897562438.1.7.6 filter(): Choose Rows
# Filter high mass proteins
proteomics_data %>%
filter(mass > 100000) %>%
select(accession, gene, mass)
#> # A tibble: 87 × 3
#> accession gene mass
#> <chr> <chr> <dbl>
#> 1 F1LMU0 F1LMU0 222850.
#> 2 Q64578 AT2A1 109409.
#> 3 D4AEH9 D4AEH9 174331.
#> 4 G3V7K1 G3V7K1 164712.
#> 5 A0A0G2K5P5 A0A0G2K5P5 187602.
#> 6 D3ZA38 D3ZA38 128936.
#> 7 Q03626 MUG1 165326
#> 8 Q8R4I6 Q8R4I6 103013.
#> 9 D3ZHA0 FLNC 290984.
#> 10 A0A096MK15 A0A096MK15 772118.
#> # ℹ 77 more rows
# Multiple conditions (AND)
proteomics_data %>%
filter(mass > 100000 & peptide_count > 100) %>%
select(accession, gene, mass, peptide_count)
#> # A tibble: 7 × 4
#> accession gene mass peptide_count
#> <chr> <chr> <dbl> <dbl>
#> 1 F1LMU0 F1LMU0 222850. 338
#> 2 Q64578 AT2A1 109409. 126
#> 3 A0A096MK15 A0A096MK15 772118. 133
#> 4 F1LRV9 F1LRV9 223400. 240
#> 5 G3V6E1 G3V6E1 219575. 173
#> 6 F1LMY4 RYR1 565491. 104
#> 7 A0A0G2K5J1 A0A0G2K5J1 563314. 102
# OR conditions
proteomics_data %>%
filter(mass > 200000 | peptide_count > 300) %>%
select(accession, gene, mass, peptide_count)
#> # A tibble: 29 × 4
#> accession gene mass peptide_count
#> <chr> <chr> <dbl> <dbl>
#> 1 F1LMU0 F1LMU0 222850. 338
#> 2 D3ZHA0 FLNC 290984. 55
#> 3 A0A096MK15 A0A096MK15 772118. 133
#> 4 F1LRV9 F1LRV9 223400. 240
#> 5 A0A0G2JU96 A0A0G2JU96 571866. 34
#> 6 M0R9L0 M0R9L0 220194. 28
#> 7 A0A0G2K7B6 A0A0G2K7B6 242460. 24
#> 8 F7F9U6 F7F9U6 517417. 16
#> 9 A0A0G2JUP3 A0A0G2JUP3 815466. 8
#> 10 G3V6E1 G3V6E1 219575. 173
#> # ℹ 19 more rows1.7.7 mutate(): Create New Columns
# Calculate log10 transformed values
proteomics_data %>%
mutate(
log10_mass = log10(mass)
) %>%
select(accession, mass, log10_mass, `2126001_029_f5`) %>%
head(3)
#> # A tibble: 3 × 4
#> accession mass log10_mass `2126001_029_f5`
#> <chr> <dbl> <dbl> <dbl>
#> 1 F1LMU0 222850. 5.35 25114306.
#> 2 G3V8V3 97288. 4.99 18519365.
#> 3 A0A0G2JSP8 43019. 4.63 897562438.1.7.8 arrange(): Sort Data
# Sort by mass (ascending)
proteomics_data %>%
arrange(mass) %>%
select(accession, gene, mass) %>%
head(3)
#> # A tibble: 3 × 3
#> accession gene mass
#> <chr> <chr> <dbl>
#> 1 P29418 ATP5E 5767.
#> 2 Q9JJW3 ATPMK 6408.
#> 3 A9UMV7 A9UMV7 6539.
# Sort descending
proteomics_data %>%
arrange(desc(mass)) %>%
select(accession, gene, mass) %>%
head(3)
#> # A tibble: 3 × 3
#> accession gene mass
#> <chr> <chr> <dbl>
#> 1 A0A0G2JUP3 A0A0G2JUP3 815466.
#> 2 A0A096MK15 A0A096MK15 772118.
#> 3 F1M1J2 F1M1J2 623402.
# Sort by multiple columns
proteomics_data %>%
arrange(desc(peptide_count), mass) %>%
select(accession, peptide_count, mass) %>%
head(3)
#> # A tibble: 3 × 3
#> accession peptide_count mass
#> <chr> <dbl> <dbl>
#> 1 F1LMU0 338 222850.
#> 2 F1LRV9 240 223400.
#> 3 G3V6E1 173 219575.1.7.9 summarize(): Calculate Statistics
# Calculate summary statistics
proteomics_data %>%
summarize(
n_proteins = n(),
mean_mass = mean(mass),
median_mass = median(mass),
sd_mass = sd(mass),
mean_intensity = mean(`2126001_029_f5`)
)
#> # A tibble: 1 × 5
#> n_proteins mean_mass median_mass sd_mass mean_intensity
#> <int> <dbl> <dbl> <dbl> <dbl>
#> 1 824 57788. 39318. 75984. 3059149.
# Multiple samples
proteomics_data %>%
summarize(
mean_s1 = mean(`2126001_029_f5`),
mean_s2 = mean(`2126001_359_f9`),
cv_s1 = sd(`2126001_029_f5`) / mean(`2126001_029_f5`) * 100
)
#> # A tibble: 1 × 3
#> mean_s1 mean_s2 cv_s1
#> <dbl> <dbl> <dbl>
#> 1 3059149. 3367563. 1102.1.7.10 group_by(): Grouped Operations
# Create groups based on mass
proteomics_data_grouped <- proteomics_data %>%
mutate(
mass_category = case_when(
mass < 50000 ~ "Low",
mass < 150000 ~ "Medium",
TRUE ~ "High"
)
)
# Calculate statistics by group
proteomics_data_grouped %>%
group_by(mass_category) %>%
summarize(
n_proteins = n(),
mean_peptides = mean(peptide_count),
mean_confidence = mean(confidence_score)
)
#> # A tibble: 3 × 4
#> mass_category n_proteins mean_peptides mean_confidence
#> <chr> <int> <dbl> <dbl>
#> 1 High 43 45.8 4136.
#> 2 Low 529 8.71 814.
#> 3 Medium 252 13.6 1176.1.7.11 Combining Operations: Pipeline
# Complete analysis pipeline
results <- proteomics_data %>%
# Filter high confidence proteins
filter(confidence_score > 10000) %>%
# Calculate mean intensity across samples
mutate(
mean_intensity = rowMeans(select(., starts_with("sample")))
) %>%
# Keep relevant columns
select(accession, gene, mass, confidence_score, mean_intensity) %>%
# Sort by mean intensity
arrange(desc(mean_intensity))
head(results)
#> # A tibble: 6 × 5
#> accession gene mass confidence_score mean_intensity
#> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 F1LMU0 F1LMU0 2.23e5 40162. NaN
#> 2 G3V8V3 G3V8V3 9.73e4 15646. NaN
#> 3 A0A0G2JSP8 A0A0G2J… 4.30e4 15567. NaN
#> 4 Q64578 AT2A1 1.09e5 14102. NaN
#> 5 P15429 ENOB 4.70e4 12564. NaN
#> 6 P05065 ALDOA 3.94e4 11642. NaN1.7.12 tidyr: Reshaping Data
Key Functions:
-
pivot_longer()- Wide to long format -
pivot_wider()- Long to wide format -
separate()- Split one column into multiple -
unite()- Combine columns
Why reshape? Different analyses and visualizations require different formats
1.7.13 Wide vs Long Format
Wide format (your current data):
accession `2126001_029_f5` `2126001_359_f9` sample_3
F1LMU0 2511430 8316460 3577492
G3V8V3 1851936 2066635 2986710Long format:
accession Sample Intensity
F1LMU0 `2126001_029_f5` 2511430
F1LMU0 `2126001_359_f9` 8316460
F1LMU0 sample_3 3577492
G3V8V3 `2126001_029_f5` 18519361.7.14 pivot_longer(): Wide to Long
# Convert to long format
proteomics_long <- proteomics_data %>%
pivot_longer(
cols = starts_with("2"),
names_to = "Sample",
values_to = "Intensity"
)
head(proteomics_long, 10)
#> # A tibble: 10 × 9
#> accession gene peptide_count unique_peptides
#> <chr> <chr> <dbl> <dbl>
#> 1 F1LMU0 F1LMU0 338 138
#> 2 F1LMU0 F1LMU0 338 138
#> 3 F1LMU0 F1LMU0 338 138
#> 4 F1LMU0 F1LMU0 338 138
#> 5 F1LMU0 F1LMU0 338 138
#> 6 F1LMU0 F1LMU0 338 138
#> 7 F1LMU0 F1LMU0 338 138
#> 8 F1LMU0 F1LMU0 338 138
#> 9 F1LMU0 F1LMU0 338 138
#> 10 F1LMU0 F1LMU0 338 138
#> # ℹ 5 more variables: confidence_score <dbl>, mass <dbl>,
#> # description <chr>, Sample <chr>, Intensity <dbl>1.7.15 Why Long Format?
Advantages for analysis:
- Easier grouping and summarizing
- Better for ggplot2 visualizations
- Facilitates statistical modeling
- Standard format for many tools
# Calculate statistics by sample
proteomics_long %>%
group_by(Sample) %>%
summarize(
mean_intensity = mean(Intensity),
median_intensity = median(Intensity),
n_proteins = n()
) %>%
head(6)
#> # A tibble: 6 × 4
#> Sample mean_intensity median_intensity n_proteins
#> <chr> <dbl> <dbl> <int>
#> 1 2126001_029_f5 3059149. 91354. 824
#> 2 2126001_068_f3 3782834. 76089. 824
#> 3 2126001_134_f11 3189430. 84892. 824
#> 4 2126001_180_f2 2687642. 129483. 824
#> 5 2126001_198_f12 2881252. 93655. 824
#> 6 2126001_270_f7 2088286. 95672. 8241.7.16 pivot_wider(): Long to Wide
# Convert back to wide format
proteomics_wide <- proteomics_long %>%
pivot_wider(
names_from = Sample,
values_from = Intensity
)
head(proteomics_wide[, 1:10])
#> # A tibble: 6 × 10
#> accession gene peptide_count unique_peptides
#> <chr> <chr> <dbl> <dbl>
#> 1 F1LMU0 F1LMU0 338 138
#> 2 G3V8V3 G3V8V3 146 113
#> 3 A0A0G2JSP8 A0A0G2JSP8 117 109
#> 4 Q64578 AT2A1 126 89
#> 5 D4AEH9 D4AEH9 87 83
#> 6 P15429 ENOB 110 83
#> # ℹ 6 more variables: confidence_score <dbl>, mass <dbl>,
#> # description <chr>, `2126001_029_f5` <dbl>,
#> # `2126001_359_f9` <dbl>, `2126001_401_f1` <dbl>1.7.17 Working with Strings
# Extract gene names
proteomics_data %>%
mutate(
# Convert to uppercase
gene_upper = toupper(gene),
# Detect pattern
is_kinase = str_detect(description, "kinase"),
# Extract words
first_word = word(description, 1)
) %>%
select(gene, description, is_kinase, first_word) %>%
head(3)
#> # A tibble: 3 × 4
#> gene description is_kinase first_word
#> <chr> <chr> <lgl> <chr>
#> 1 F1LMU0 Myosin heavy chain 4 FALSE Myosin
#> 2 G3V8V3 Alpha-1,4 glucan phosphor… FALSE Alpha-1,4
#> 3 A0A0G2JSP8 Creatine kinase TRUE Creatine1.7.18 Handling Missing Values
# Create data with missing values
data_with_na <- proteomics_data %>%
mutate(
`2126001_029_f5` = ifelse(`2126001_029_f5` < quantile(`2126001_029_f5`, 0.2),
NA, `2126001_029_f5`)
)
# Check missing values
data_with_na %>%
summarize(
n_total = n(),
n_missing_s1 = sum(is.na(`2126001_029_f5`)),
percent_missing = mean(is.na(`2126001_029_f5`)) * 100
)
#> # A tibble: 1 × 3
#> n_total n_missing_s1 percent_missing
#> <int> <int> <dbl>
#> 1 824 165 20.0
# Remove rows with NA
data_with_na %>%
filter(!is.na(`2126001_029_f5`)) %>%
nrow()
#> [1] 659
# Replace NA with value
data_with_na %>%
mutate(`2126001_029_f5` = replace_na(`2126001_029_f5`, 0)) %>%
head(3)
#> # A tibble: 3 × 19
#> accession gene peptide_count unique_peptides
#> <chr> <chr> <dbl> <dbl>
#> 1 F1LMU0 F1LMU0 338 138
#> 2 G3V8V3 G3V8V3 146 113
#> 3 A0A0G2JSP8 A0A0G2JSP8 117 109
#> # ℹ 15 more variables: confidence_score <dbl>, mass <dbl>,
#> # description <chr>, `2126001_029_f5` <dbl>,
#> # `2126001_359_f9` <dbl>, `2126001_401_f1` <dbl>,
#> # `2126001_180_f2` <dbl>, `2126001_416_f6` <dbl>,
#> # `2126001_422_f10` <dbl>, `2126001_068_f3` <dbl>,
#> # `2126001_134_f11` <dbl>, `2126001_270_f7` <dbl>,
#> # `2126001_198_f12` <dbl>, `2126001_312_f8` <dbl>, …1.7.19 Hands-On Exercise
Task: Transform and analyze your proteomics data
# 1. Load your data
my_data <- read_excel("your_data.xlsx")
# 2. Filter and calculate
high_quality <- my_data %>%
filter(confidence_score > 10000, peptide_count > 50) %>%
mutate(mean_intensity = rowMeans(select(., starts_with("sample"))))
# 3. Convert to long format
long_data <- my_data %>%
pivot_longer(cols = starts_with("sample"),
names_to = "Sample",
values_to = "Intensity")
# 4. Calculate CV per protein
cv_data <- long_data %>%
group_by(accession) %>%
summarize(mean_int = mean(Intensity, na.rm = TRUE),
sd_int = sd(Intensity, na.rm = TRUE),
cv = (sd_int / mean_int) * 100)1.8 Day 1 Summary
Today you learned:
1.8.1 Homework
- Install all required packages for Day 2
- Practice writing functions for data manipulation
- Explore the built-in datasets in R (use
data()to see available datasets)
# Install packages for Day 2
install.packages(c("ggplot2", "dplyr", "tidyr", "pheatmap"))
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("limma", "vsn"))1.9 Additional Resources
- R for Data Science by Hadley Wickham
- RStudio Cheat Sheets
- Stack Overflow for questions